iCON Education HYD, 79930 92826, 73309 72826JEE Main 2021 (Online) 27th August Morning Shift
200 mL of 0.2 M HCl is mixed with 300 mL of 0.1 M NaOH. The molar heat of neutralization of this reaction is $-$57.1 kJ. The increase in temperature in $^\circ$C of the system on mixing is x $\times$ 10$-$2. The value of x is ___________. (Nearest integer)
[Given : Specific heat of water = 4.18 J g$-$1 K$-$1, Density of water = 1.00 g cm$-$3]
[Assume no volume change on mixing)
Correct Answer: 82
Explanation:
$\Rightarrow$ Millimoles of HCl = 200 $\times$ 0.2 = 40
$\Rightarrow$ Millimoles of NaOH = 300 $\times$ 0.1 = 30
iCON Education HYD, 79930 92826, 73309 72826JEE Main 2021 (Online) 26th August Evening Shift
For water $\Delta$vap H = 41 kJ mol$-$1 at 373 K and 1 bar pressure. Assuming that water vapour is an ideal gas that occupies a much larger volume than liquid water, the internal energy change during evaporation of water is ___________ kJ mol$-$1
The magnitude of lattice enthalpy of KCl in kJ mol$-$1 is 718 (Nearest integer).
2021
JEE Mains
Numerical
iCON Education HYD, 79930 92826, 73309 72826JEE Main 2021 (Online) 27th July Evening Shift
When 400 mL of 0.2 M H2SO4 solution is mixed with 600 mL of 0.1 M NaOH solution, the increase in temperature of the final solution is __________ $\times$ 10$-$2 K. (Round off to the nearest integer).
iCON Education HYD, 79930 92826, 73309 72826JEE Main 2021 (Online) 25th July Evening Shift
A system does 200 J of work and at the same time absorbs 150 J of heat. The magnitude of the change in internal energy is ____________ J. (Nearest integer)
Correct Answer: 50
Explanation:
w = $-$200 J, q = +150 : $\Delta$U = q + w
$\Delta$U = 150 $-$ 200 = $-$50 J
Magnitude = 50 J = |$\Delta$U |
2021
JEE Mains
Numerical
iCON Education HYD, 79930 92826, 73309 72826JEE Main 2021 (Online) 25th July Morning Shift
At 298 K, the enthalpy of fusion of a solid (X) is 2.8 kJ mol$-$1 and the enthalpy of vaporisation of the liquid (X) is 98.2 kJ mol$-$1. The enthalpy of sublimation of the substance (X) in kJ mol$-$1 is _____________. (in nearest integer)
Correct Answer: 101
Explanation:
The enthalpy of sublimation is the total amount of energy required to convert a solid directly into a gas. This can be calculated by summing the enthalpy of fusion (solid to liquid) and the enthalpy of vaporization (liquid to gas). Mathematically, this relationship is represented as:
Therefore, the enthalpy of sublimation of the substance (X) is approximately 101 kJ mol$-1$.
2021
JEE Mains
Numerical
iCON Education HYD, 79930 92826, 73309 72826JEE Main 2021 (Online) 22th July Evening Shift
If the standard molar enthalpy change for combustion of graphite powder is $-$2.48 $\times$ 102 kJ mol$-$1, the amount of heat generated on combustion of 1 g of graphite powder is ___________ kJ. (Nearest integer)
Correct Answer: 21
Explanation:
1 mol graphite = 12 gm C
For 1 g of graphite = ${{248} \over {12}}$ = 20.67 kJ/gm heat evolved.
2021
JEE Mains
Numerical
iCON Education HYD, 79930 92826, 73309 72826JEE Main 2021 (Online) 20th July Evening Shift
For a given chemical reaction A $\to$ B at 300 K the free energy change is $-$49.4 kJ mol$-$1 and the enthalpy of reaction is 51.4 kJ mol$-$1. The entropy change of the reaction is _____________ JK$-$1 mol$-$1.
iCON Education HYD, 79930 92826, 73309 72826JEE Main 2021 (Online) 16th March Evening Shift
At 25$^\circ$C, 50 g of iron reacts with HCl to form FeCl2. The evolved hydrogen gas expands against a constant pressure of 1 bar. The work done by the gas during this expansion is _________ J. (Round off to the Nearest Integer).
[Given : R = 8.314 J mol$-$1 K$-$1. Assume, hydrogen is an ideal gas] [Atomic mass of Fe is 55.85 u]
Correct Answer: 2218
Explanation:
$Fe + 2HCl \to FeC{l_2} + {H_2}$
${{50} \over {55.85}}moles$
Moles of Fe = ${{50} \over {55.85}}moles$ = Moles of H2
iCON Education HYD, 79930 92826, 73309 72826JEE Main 2021 (Online) 26th February Morning Shift
For a chemical reaction A + B ⇌ C + D
(${\Delta _r}{H^\Theta }$ = 80 kJ mol$-$1) the entropy change ${\Delta _r}{S^\Theta }$ depends on the temperature T (in K) as ${\Delta _r}{S^\Theta }$ = 2T (J K$-$1mol$-$1).
Minimum temperature at which it will become spontaneous is ___________ K. (Integer)
The minimum temperature to make it spontaneous is 200 K.
2021
JEE Mains
Numerical
iCON Education HYD, 79930 92826, 73309 72826JEE Main 2021 (Online) 26th February Morning Shift
An exothermic reaction X $ \to $ Y has an activation energy 30 kJ mol$-$1. If energy change $\Delta$E during the reaction is $-$20 kJ, then the activation energy for the reverse reaction in kJ is ___________. (Integer answer)
Correct Answer: 50
Explanation:
X $ \to $ Y
$\Delta $E = (Ea)f
– (Ea)b
$ \Rightarrow $ – 20 = 30 – (Ea)b
$ \Rightarrow $ (Ea)b = 50 kJ
2021
JEE Mains
Numerical
iCON Education HYD, 79930 92826, 73309 72826JEE Main 2021 (Online) 25th February Evening Shift
Five moles of an ideal gas at 293 K is expanded isothermally from an initial pressure of 2.1 MPa to 1.3 MPa against at constant external pressure 4.3 MPa. The heat transferred in this process is _________ kJ mol$-$1. (Rounded off to the nearest integer) [Use R = 8.314 J mol$-$1K$-$1]
Correct Answer: 15
Explanation:
The gas performs isothermal irreversible work (W).
iCON Education HYD, 79930 92826, 73309 72826JEE Main 2021 (Online) 25th February Morning Shift
The reaction of cyanamide, NH2CN(s) with oxygen was run in a bomb calorimeter and $\Delta$U was found to be $-$742.24 kJ mol$-$1. The magnitude of $\Delta$H298 for the reaction
iCON Education HYD, 79930 92826, 73309 72826JEE Main 2021 (Online) 25th February Morning Shift
The ionization enthalpy of Na+ formation from Na(g) is 495.8 kJ mol$-$1, while the electron gain enthalpy of Br is $-$325.0 kJ mol$-$1. Given the lattice enthalpy of NaBr is $-$728.4 kJ mol$-$1. The energy for the formation of NaBr ionic solid is ($-$) ____________ $\times$ 10$-$1 kJ mol$-$1.
iCON Education HYD, 79930 92826, 73309 72826JEE Main 2021 (Online) 24th February Evening Shift
Assuming ideal behaviour, the magnitude of log K for the following reaction at 25$^\circ$C is x $\times$ 10$-$1. The value of x is ______________. (Integer answer)
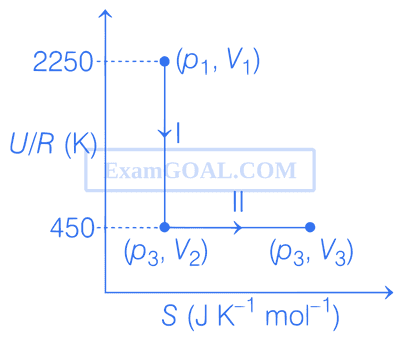
One mole of an ideal gas at 900 K, undergoes two reversible processes, I followed by II, as shown below. If the work done by the gas in the two processes are same, the value of $\ln {{{V_3}} \over {{V_2}}}$ is _________.
(U : internal energy, S : entropy, p : pressure, V : volume, R : gas constant)
(Given : molar heat capacity at constant volume, CV,m of the gas is ${5 \over 2}$R)
Correct Answer: 10
Explanation:
For process 1, entropy is constant thus, q is constant
$\therefore$ ${W_I} = \Delta U = n{C_{V,m}}\Delta T$
An ideal gas undergoes a reversible isothermal expansion from state I to state II followed by a reversible adiabatic expansion from state II to state III. The correct plot(s) representing the changes from state I to state III is (are)
(p : pressure, V : volume, T : temperature, H : enthalpy, S : entropy)
A.
B.
C.
D.
Correct Answer: A,B,D
Explanation:
From state I to II Reversible isothermal expansion takes
place. So, following changes take place,
● pressure decreases.
● Volume increases.
● Temperature remains constant.
● Enthalpy, H remains constant.
● Entropy, S for expansion increases.
So, all options follows the above mentioned conditions, so all
graphs are correct for state I and II.
From state II to III Reversible adiabatic expansion takes place.
So, following changes take place.
● pressure decreases.
● Volume increases.
● Temperature decreases.
● Enthalpy, H decreases.
● Entropy, S remains constant.
● H increases instead of decreasing, so only option (c) is
incorrect.
All other options, i.e., (a), (b) and (d) follows the above
mentioned conditions.
Therefore, correct graphical representations are (a), (b) and (d).
When an ideal gas expands isothermally from $5 \mathrm{~m}^3$ to $10 \mathrm{~m}^3$ at $25^{\circ} \mathrm{C}$ against a constant pressure of $10^7 \mathrm{~Nm}^{-2}$, then the work done on the gas is
A.
$-100 \mathrm{~MJ}$
B.
$-50 \mathrm{~MJ}$
C.
$-0.5 \mathrm{~MJ}$
D.
$-10^5 \mathrm{~MJ}$
Correct Answer: B
Explanation:
Work done on isothermal irreversible expansion for ideal gas
Find the approximate value of $(\Delta H-\Delta U)$ in $\mathrm{Jmol}^{-1}$, for the formation of CO from its elements at $298 \mathrm{~K} .\left(R=8.314 \mathrm{JK}^{-1} \mathrm{~mol}^{-1}\right)$
For the reaction, $\mathrm{H}_2 \mathrm{O}(l) \longrightarrow \mathrm{H}_2 \mathrm{O}(\mathrm{g})$ at $T=100^{\circ} \mathrm{C}$ and $p=1 \mathrm{~atm}$, choose the correct option.
Two flasks $A$ and $B$ have equal volumes. $A$ is maintained at $300 \mathrm{~K}$ and $B$ at $600 \mathrm{~K}$. Equal masses of $\mathrm{H}_2$ and $\mathrm{CO}_2$ are taken in flasks $A$ and $B$ respectively. Find the ratio of total KE of gases in flask $A$ to that of $B$.
A.
1 : 2
B.
11 : 1
C.
33 : 2
D.
55 : 7
Correct Answer: B
Explanation:
Formula of kinetic energy $=\frac{3 n R T}{2}$
Total kinetic energy expression for $A$ and $B$ flask,
$\mathrm{KE}_A=\frac{3 n_A R T_A}{2} \text { and } \mathrm{KE}_B=\frac{3 n_B R T_B}{2} .$ ...... (i)
Let us consider common mass $=m$
$n_A=\frac{m}{2} \quad \text { and } \quad n_B=\frac{m}{44}$
If a chemical reaction is known to be
non-spontaneous at 298 K but spontaneous
at 350 K, then which among the following
conditions is true for the reaction?
A.
$\Delta G=-v e, \Delta H=-v e, \Delta S=+ ve$
B.
$\Delta G=+v e, \Delta H=+v e, \Delta S=+v e$
C.
$\Delta G=-v e, \Delta H=+v e, \Delta S=+v e$
D.
$\Delta G=+ ve, \Delta H=+ ve, \Delta S=-v e$
Correct Answer: C
Explanation:
According to Gibb's free energy, $\Delta G=\Delta H-T \Delta S$
Reaction is non spontaneous i.e. $\Delta G > 0$ at $298 \mathrm{~K}$.
As the temperature increases from $298 \mathrm{~K}$ to $350 \mathrm{~K}$.
The reaction becomes spontaneous i.e. $\Delta G < 0$.
$\begin{gathered}
\Delta G=\Delta H-T \Delta S \\
\Delta G < 0\end{gathered}$
When $\Delta H$ and $\Delta S$ are + ve then, $\Delta G$ will be positive till $\Delta H > T \Delta S$.
When the temperature is increased further $\Delta H$ becomes less than $T \Delta S$ i.e.
$\Delta H < T \Delta S$ and $\Delta G$ becomes $-$ve.
Thus, reaction becomes spontaneous.
2020
JEE Mains
MCQ
iCON Education HYD, 79930 92826, 73309 72826JEE Main 2020 (Online) 6th September Evening Slot
For a reaction,
4M(s) + nO2(g) $ \to $ 2M2On(s)
the free energy change is plotted as a function
of temperature. The temperature below which
the oxide is stable could be inferred from the
plot as the point at which :
A.
the free energy change shows a change
from negative to positive value
B.
the slope changes from positive to negative
C.
the slope changes from negative to positive
D.
the slope changes from positive to zero
Correct Answer: A
Explanation:
$\Delta $G = $\Delta $H – T$\Delta $S
$\Delta $G = –ve (stable oxide)
$\Delta $G = +ve (unstable oxide)
2020
JEE Mains
MCQ
iCON Education HYD, 79930 92826, 73309 72826JEE Main 2020 (Online) 5th September Evening Slot
Lattice enthalpy and enthalpy of solution of NaCl are 788 kJ mol–1, and 4 kJ mol–1, respectively.
The hydration enthalpy of NaCl is :
A.
–780 kJ mol–1
B.
–784 kJ mol–1
C.
780 kJ mol–1
D.
784 kJ mol–1
Correct Answer: B
Explanation:
$\Delta $Hsol = Lattice enthalpy + $\Delta $Hhyd
$ \Rightarrow $ 4 = 788 + $\Delta $Hhyd
$ \Rightarrow $ $\Delta $Hhyd = –784 kJ mol–1
2020
JEE Mains
MCQ
iCON Education HYD, 79930 92826, 73309 72826JEE Main 2020 (Online) 4th September Evening Slot
Five moles of an ideal gas at 1 bar and 298 K
is expanded into vacuum to double the volume.
The work done is :
A.
Zero
B.
-RT $\ln {{{V_2}} \over {{V_1}}}$
C.
CV (T2 – T1)
D.
– RT (V2 – V1)
Correct Answer: A
Explanation:
As the expansion is done in vacuum that is in absence of pext so
pext = 0
$ \therefore $ W = - pext$\Delta $V
= 0
2020
JEE Mains
MCQ
iCON Education HYD, 79930 92826, 73309 72826JEE Main 2020 (Online) 4th September Evening Slot
The process that is NOT endothermic in nature
is :
A.
Ar(g) + e- $ \to $ Ar-(g)
B.
H(g) + e- $ \to $ H-(g)
C.
Na(g) $ \to $ Na+(g) + e-
D.
O-(g) + e- $ \to $ O2-(g)
Correct Answer: B
Explanation:
H(g) + e- $ \to $ H-(g) is exothermic
rest of all endothermic process.
2020
JEE Mains
MCQ
iCON Education HYD, 79930 92826, 73309 72826JEE Main 2020 (Online) 4th September Morning Slot
For one mole of an ideal gas, which of these
statements must be true?
(a) U and H each depends only on temperature
(b) Compressibility factor z is not equal to 1
(c) CP, m – CV, m = R
(d) dU = CVdT for any process
A.
(a), (c) and (d)
B.
(a) and (c)
C.
(c) and (d)
D.
(b), (c) and (d)
Correct Answer: A
Explanation:
For 1 mole of ideal gas :
1. Both internal energy (U) and Enthalpy (H)
depends on temperature
2. Compressibility factor Z = 1
3. CP, m – CV, m = R
4. dU = CVdT for all process
2020
JEE Mains
MCQ
iCON Education HYD, 79930 92826, 73309 72826JEE Main 2020 (Online) 9th January Evening Slot
The true statement amongst the following is :
A.
S is a function of temperature but $\Delta $S is not
a function of temperature.
B.
Both S and $\Delta $S are not functions of
temperature.
C.
Both $\Delta $S and S are functions of temperature.
D.
S is not a function of temperature but $\Delta $S is
a function of temperature.
Correct Answer: C
Explanation:
$\Delta S = \int {{{d{q_{rev}}} \over T}} $
S = Kln(w)
Both entropy and change in entropy are
function of temperature.
2020
JEE Mains
MCQ
iCON Education HYD, 79930 92826, 73309 72826JEE Main 2020 (Online) 9th January Morning Slot
If enthalpy of atomisation for Br2(1) is x kJ/mol
and bond enthalpy for Br2 is y kJ/mol, the
relation between them :
A.
does not exist
B.
is x < y
C.
is x > y
D.
is x = y
Correct Answer: C
Explanation:
$ \therefore $ $\Delta $Hatomisation = $\Delta $HVap + Bond Energy
$ \Rightarrow $ x = $\Delta $HVap + y
$ \Rightarrow $ x $>$ y
2020
JEE Mains
Numerical
iCON Education HYD, 79930 92826, 73309 72826JEE Main 2020 (Online) 5th September Evening Slot
For a dimerization reaction,
2A(g) $ \to $ A2(g)
at 298 K, $\Delta $Uo
= –20 kJ mol–1, $\Delta $So
= –30
JK–1 mol–1, then the $\Delta $Go
will be _____ J.
iCON Education HYD, 79930 92826, 73309 72826JEE Main 2020 (Online) 2nd September Evening Slot
The heat of combustion of ethanol into carbon
dioxide and water is – 327 kcal at constant
pressure. The heat evolved (in cal) at constant
volume and 27oC (if all gases behave ideally) is
(R = 2 cal mol–1 K–1) ________.
iCON Education HYD, 79930 92826, 73309 72826JEE Main 2020 (Online) 8th January Evening Slot
At constant volume, 4 mol of an ideal gas when
heated from 300 K to 500K changes its internal
energy by 5000 J. The molar heat capacity at
constant volume is _______.
Correct Answer: 6.25
Explanation:
$\Delta $U = nCv$\Delta $T
$ \Rightarrow $ 5000 = 4 × Cv(500 – 300)
$ \Rightarrow $ Cv = 6.25 JK–1mol–1
2020
JEE Mains
Numerical
iCON Education HYD, 79930 92826, 73309 72826JEE Main 2020 (Online) 8th January Morning Slot
The magnitude of work done by a gas that undergoes a reversible expansion along the path ABC shown in
the figure is _______.
Correct Answer: 48
Explanation:
Work done = Area covered by the diagram
= $1\over2$ × (sum of parallel sides) × height
= $1\over2$ × (10+6) × 6
= $1\over2$ × 16 × 6
= 48 Joule
Note : Here pressure difference = 8 - 2 = 6 Pa and volume difference = 12 - 2 = 10
2020
JEE Mains
Numerical
iCON Education HYD, 79930 92826, 73309 72826JEE Main 2020 (Online) 7th January Evening Slot
The standard heat of formation $\left( {{\Delta _f}H_{298}^0} \right)$ of ethane (in kj/mol), if the heat of combustion of
ethane, hydrogen and graphite are - 1560, -393.5 and -286 Kj/mol, respectively is :
Tin is obtained from cassiterite by reduction with coke. Use the data given below to determine the minimum temperature (in K) at which the reduction of cassiterite by coke would take place.
At $298K:{\Delta _f}H^\circ [Sn{O_2}(s)] = - 581.0$ mol-1,
But, it is not applicable for irreversible process which are carried out very fast. So, work done is calculated assuming final pressure remains constant throughout the process. Thus, statement (a), (b) and (c) correct while statement (d) is incorrect.
Which of the following statements regarding the first law of thermodynamics is correct?
A.
The energy of the isolated system plus the energy of the surrounding is constant.
B.
The energy of the isolated system minus the energy of the surrounding is constant.
C.
The energy of an isolated system is constant.
D.
The energy of an isolated system varies.
Correct Answer: C
Explanation:
∵ According to first law of thermodynamics, the total energy of an isolated system remains constant, through it may change from one form to another. Thus, statement (c) is correct. Hence, option (c) is the correct answer.
What will be the $\Delta U$ value, when one mole of oxygen $\left(\mathrm{O}_2\right)$ is going from $-20^{\circ} \mathrm{C}$ to $40^{\circ} \mathrm{C}$ at constant volume? (Molar heat capacity for oxygen $\simeq 20.8 \mathrm{~J} \mathrm{~mol}^{-1} \mathrm{~K}^{-1}$ )
A.
2496 J
B.
20.8 J
C.
416 J
D.
1248 J
Correct Answer: D
Explanation:
∵ Given, Molar heat capacity for oxygen
$ \begin{aligned} & =20.8 \mathrm{~J} \mathrm{~mol}^{-1} \mathrm{~K}^{-1} \\ Q & =n C \Delta T \end{aligned} $
where, $Q=$ value of $\Delta U$ in J
$ \begin{aligned} n & =\text { number of moles }(=1) \\ C & =\text { heat capacity } \\ \Delta T & =\text { temperature } \\ Q & =1 \times 20.8 \times 60 \end{aligned} $
Thus, $\Delta T=(-) 20$ to $(+) 40=60^{\circ} \mathrm{C}$
Hence, for one mole of oxygen, value of $\Delta U=60 \times 20.8=1248 \mathrm{~J}$ Hence, option (d) is the correct answer.